Overview and Impact to Werfen
In Vitro Diagnostic Regulation (IVDR)
What is IVDR?
IVDR is a new European Regulation (EU 2017/746), applicable to all in vitro diagnostic (IVD) medical devices, current and new. This Regulation will replace the current European Directive IVDD (Directive 98/79/EC) for IVD devices.
Territories Impacted
As a European regulation, IVDR will be effective in all European Union (EU) member states and European Free Trade Agreement (EFTA) states.
EU member states:
Austria, Belgium, Bulgaria, Croatia, Cyprus, Czech Republic, Denmark, Estonia, Finland, France, Germany, Greece, Hungary, Ireland, Italy, Latvia, Lithuania, Luxembourg, Malta, Netherlands, Poland, Portugal, Romania, Slovakia, Slovenia, Spain, and Sweden.
EFTA states:
Iceland, Liechtenstein, Norway, and Switzerland
Timeline for IVDR Implementation
May 26, 2017: IVDR first published, 5-year transition period begins
May 26, 2022: IVDR in full effect
- IVD devices already in inventory in EU/EFTA territories can continue to be sold or distributed in a sell-off period
- Analyzers not IVDR-compliant, but sold prior to May 26, 2022, can remain in the market until end-of-life
May 26, 2025: Sell-off period ends for all IVD devices; from this date forward only IVDR compliant devices can be sold or distributed.
IVDR Classification
IVDR re-classifies IVD medical devices, based on new criteria for clinical risk associated with use.
| Class | Includes |
|---|---|
| Class A (lowest risk) | Analyzers, reaction cuvettes, washing solutions or other accessories |
| Class B (moderate risk) | Reagents, controls or calibrators (e.g., used in clinical chemistry, autoimmunity, coagulation) |
| Class C (high risk) | Tests used in emergency settings for the monitoring of critical parameters (e.g., D-dimer, anticoagulant therapy monitoring tests, prenatal screening, cardiac markers, tumor markers, self-testing for Blood Glucose, serology tests to detect specific viruses, such as HPV, CMV, Rubella) |
| Class D (highest risk) | Tests used for the determination of blood groups or infection disease (e.g., HIV and hepatitis B/C/D, SARS-CoV-2) |
| Near-patient tests (a new designation) | Devices to be used in a near-patient setting, only by a healthcare professional, not by a patient. Near-patient tests are performed by healthcare professionals, and self-tests are performed by lay persons. |
Additional information on classifications: https://ec.europa.eu/health/sites/health/files/md_sector/docs/md_mdcg_2020_guidance_classification_ivd-md_en.pdf
IVDR CE Mark Requirements
IVDR Conformitè Europëenne (CE) mark compliance is the seal of approval for IVD products, as effective and safe for use. Before placing a device in the European market, a manufacturer must obtain the CE mark, by completing the conformity assessment of the device with a Notified Body. This procedure differs by device classification:
- Class A requirements: Conformity and self-certification for CE mark, performed by the manufacturer.
- Classes B, C, D requirements: Conformity and certification for CE mark, performed by an independent Notified Body, with approval required prior to implementation and distribution.
The process to obtain IVDR compliance includes:
- Updating technical documents, including analytical, clinical performance, clinical evidence, stability, risk, manufacturing procedures, and other relevant business processes.
- Verification of technical and scientific competences required by the Notified Body.
- Review of technical documents to ensure compliance with IVDR requirements. Higher risk devices have stricter conformity assessments.
- Releasing and signing the CE Declaration of Conformity, upon meeting all IVDR requirements, and release of the CE mark certificate by the Notified Body
- Updating packaging, labelling and instructions for use (in Classes B, C or D) to reflect conformity, including a new identification number (xxxx) of the Notified Body, placed in proximity to the CE mark symbol (shown right).
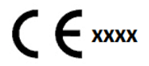
- No changes to design or manufacturing processes.
Refer to European Commission website for details: https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=35
Impact to Customers
Once the IVDR is active, a central European database, Eudamed, will be implemented. Eudamed will be accessible and will house specific, related documents and information.
Example: Certificates, and occurrences of field safety corrective actions and serious adverse events, will be published in Eudamed.
IVDR aims to ensure the quality, safety and reliability of IVD medical devices in the European market. IVDR will increase the transparency of information available publicly.
GEM Premier and ROTEM systems only: Printed Instructions for Use are now part of IVDR labeling requirements for near-patient tests. Printed copies of the Operators Manuals for GEM Premier and ROTEM systems will be provided to users, prior to IVDR implementation.